Protéomique
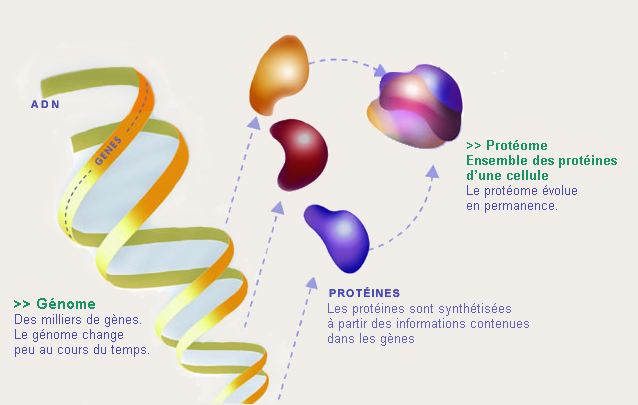
La protéomique sert à désigner la science qui étudie les protéomes, c'est-à-dire la totalité des protéines d'une cellule, organites, tissu, organe ou organisme à un moment donné et sous des conditions données.

Catégories :
Protéine - Discipline de la biologie
Recherche sur Google Images :
Source image : interstices.info Cette image est un résultat de recherche de Google Image. Elle est peut-être réduite par rapport à l'originale et/ou protégée par des droits d'auteur. |
Définitions :
- Etude, qui se voudrait complète, des produits des gènes, les protéines, présentes dans une cellule ou un tissu, dans un environnement donné.... (source : genopole)
La protéomique sert à désigner la science qui étudie les protéomes, c'est-à-dire la totalité des protéines d'une cellule, organites, tissu, organe ou organisme à un moment donné et sous des conditions données.
Dans la pratique, la protéomique à pour but de identifier les protéines extraites d'une culture cellulaire, d'un tissu ou d'un fluide biologique, leur localisation dans les compartiments cellulaires, leurs modifications post-traductionnelles mais aussi leur quantité. Elle sert à quantifier les variations de leur taux d'expression selon le temps, de leur environnement, de leur état de développement, de leur état physiologique et pathologique, de l'espèce d'origine. Elle étudie aussi les interactions que les protéines ont avec d'autres protéines, avec l'ADN ou l'ARN, avec des substances. La protéomique fonctionnelle étudie les fonctions de chaque protéine. La protéomique étudie enfin la structure primaire, secondaire et tertiaire des protéines.
Histoire de la protéomique
Le terme de protéomique est récent. Il a été utilisé pour la première fois dans une publication scientifique en 1997 par James P. [1] dans son article Identification des protéines dans l'ère post-génomique : l'ascension rapide de la protéomique. Il dérive de protéome, terme découvert en 1995, par ressemblance avec génomique qui dérive lui-même du terme génome, la totalité des gènes d'un organisme. L'analyse protéomique est une étude dynamique. Un seul génome peut conduire à différents protéomes suivant les étapes du cycle cellulaire, de la différenciation, de la réponse à différents signaux biologiques ou physiques, de l'état physiopathologique, ... Le protéome reflète les répercussions de ces événements cellulaires au niveau tant traductionnel que post-traductionnel. De ce point de vue, seule une analyse protéique directe peut donner une image globale des dispositifs biomoléculaires dans leur complexité.
Les scientifiques se sont intéressés aux protéines bien avant l'apparition de la protéomique. Dès 1833, Anselme Payen et Jean-François Persoz isolèrent d'un extrait de malt une enzyme capable de catalyser la dégradation de l'amidon en sucre. en 1965, André Lwoff, François Jacob et Jacques Monod reçurent le prix Nobel de médecine «pour leurs recherches sur la manière dont la production des enzymes est réglée au niveau des gènes»[2], publiées en 1961).
De nombreuses techniques, toujours beaucoup utilisées, ont été développées.
La technique d'électrophorèse a été développée en 1892 par S. E. Linder et H. Picton. Le principe de la chromatographie date de 1861 par Friedrich Goppelsröder.
// mettre une fresque reprenant la chronologie de l'étude des protéines
Depuis une dizaine d'années, la protéomique est devenue une science à part entière, avec ses techniques propres et ses méthodes.
Elle a emprunté de nombreuses technologies, jusque là utilisées dans d'autres disciplines, et les a appliquées à à l'étude des protéines. On peut citer par exemple l'utilisation de la spectrométrie de masse, provenant de la physique et de l'analyse chimique, dans l'identification des protéines, dans la quantification de l'expression des protéines, dans la localisation de peptides dans un tissu, dans la recherche de biomarqueurs spécifiques de pathologies.
Pourquoi le protéome ?
Prenant en compte l'existence de la formidable somme d'informations issues de l'analyse génomique, on peut se demander pourquoi des projets d'analyse protéomique fréquemment complexes à mettre en œuvre et coûteux doivent s'y surajouter et quelles informations spécifiques en attendre. Pour comprendre en quoi l'étude du protéome apparaît actuellement comme une obligation, il suffit de revenir sur les événements clés qui vont conduire du stock d'informations que forme le génome à la production des molécules qui vont déterminer et réguler la vie cellulaire, les protéines. En principe, la séquence de chaque gène sera transcrite (ou non) en un ARN messager (ARN-m), lui-même traduit en protéine. En réalité, les gènes des cellules eucaryotes sont fréquemment morcelés et contiennent des régions (introns) absentes de l'ARN-m. Une transcription partielle sera permise par l'épissage d'un ARN précurseur, copie du gène, pouvant donner naissance à différents ARN-m, chacun de ceux-ci pouvant aboutir à plusieurs protéines. On peut par conséquent avancer une série de raisons plaidant en faveur du développement de l'analyse protéomique :
• Non seulement l'identification mais l'estimation des taux de protéines sont principales pour obtenir une image complète de processus biologiques divers. Cependant, l'abondance des protéines au sein de la cellule n'est pas uniquement régulée à un niveau transcriptionnel, ainsi qu'aux niveaux traductionnels et post-traductionnels, de telle sorte qu'aucune relation simple ne peut être établie entre taux d'ARN-m et de protéines. Le fait qu'un gène unique, et même un ARN-m unique, puisse conduire à plusieurs protéines différentes par leur (s) fonction (s) rend hasardeuse cette corrélation. D'autre part, certaines protéines ont une durée de vie longue, c'est-à-dire que même synthétisées à faible vitesse elles peuvent s'accumuler dans la cellule en demeurant fonctionnelles, tandis que d'autres à durée de vie brève sont rapidement éliminées. Donc, même si leur synthèse est rapide et elles se retrouveront à un faible taux.
• Une même protéine pourra selon l'état cellulaire (différenciation, prolifération, apoptose) se retrouver dans un compartiment donné (cytoplasme, noyau, mitochondrie) ou être secrétées par la cellule. Sans l'analyse du protéome, une modification de localisation de la protéine indispensable à son activité biologique passera inaperçue.
• La plupart des protéines ne parviennent à leur forme biologiquement active qu'suite à étapes de maturation co- et post-traductionnelles telles que glycosylation, phosphorylation, déamination, ... Ces modifications confèrent fréquemment à la protéine sa fonctionnalité. Elles sont aussi les indicateurs de l'état de la machinerie cellulaire.
Finalement, les résultats du projet de séquençage du génome humain forment un argument définitif pour l'obligation d'une étude approfondie du protéome. La découverte étonnante que ce génome contienne nettement moins de gènes que prédits démontre l'importance des protéines comme acteurs centraux des processus biologique
Différentes approches de la protéomique
Les méthodologies mises en œuvre pour l'analyse du protéome peuvent schématiquement être scindées en deux grands groupes : dans le premier peuvent être regroupées les méthodes expérimentales réalisées dans le «laboratoire réel» (wet laboratory), reposant essentiellement sur les techniques de séparation et d'analyse des protéines. Un second groupe de méthodes, mises en œuvre dans le «laboratoire virtuel» (dry laboratory), fait appel à l'analyse d'images ainsi qu'à la bio-informatique. Laboratoires réel et virtuel sont mis à contribution lors des principales étapes de l'analyse.
Dans la pratique, les protéines sont en premier lieu extraites d'une population cellulaire ou d'un tissu, puis scindées avant d'être identifiées.
Extraction
La première étape consiste le plus souvent à extraire les protéines d'un échantillon biologique. Cette étape est cruciale : une mauvaise extraction peut produire la dégradation des protéines et compliquer, voir rendre impossible, l'identification des protéines. Les protéines membranaires, comportant de nombreux acides aminées hydrophobes et par conséquent peu soluble, restent complexes à étudier.
Certaines techniques ne nécessitent pas d'extraire les protéines du tissu étudié. Dans l'immunolocalisation, le tissu est fixé puis découpé en fines lamelles de quelques microns d'épaisseur. Les protéines sont ensuite détectées in situ par des anticorps marqués. Dans l'imagerie par spectrométrie de masse, des coupes de tissus sont analysées directement par un spectromètre de masse de type MALDI-TOF.
Pour simplifier l'analyse, l'extraction est fréquemment réalisée en éliminant les modifications post-traductionnelles. Seule la structure primaire des protéines, c'est-à-dire leurs séquences, est conservée. Mais si le sujet de l'analyse est l'étude de ces modifications post-traductionnelles, il convient de prendre les précautions nécessaires pour les garder.
Séparation
La seconde étape sert à séparer les protéines selon leurs caractéristiques physiques ou chimiques ou selon leurs affinités pour un ligand.
L'électrophorèse sépare les protéines dans un gel polyacrylamide selon leur taille quand elles sont soumises à un champ électrique. La méthode d'électrophorèse de référence pour la protéomique est l'électrophorèse bidimensionnelle (2-DE).
La chromatographie utilise la différence d'affinité des protéines entre une phase mobile et une phase stationnaire.
Principe de la séparation des protéines par électrophorèse bidimensionnelle
L'électrophorèse bidimensionnelle autorise partir de mélanges protéiques complexes de séparer et visualiser des centaines ou alors des milliers de protéines sous forme de taches ou «spots». La résolution obtenue est suffisante pour mettre en évidence la présence d'isoformes.
Son principe consiste à effectuer tout d'abord une séparation des protéines selon leur charge (isoélectrofocalisation, IEF), suivie d'une séparation orthogonale, selon la taille. La résolution de la première dimension est de l'ordre de 0.01 unités pH.
Les gels obtenus sont ensuite colorés, puis numérisés. Le résultat est une semi-quantification.
Recherche des protéines d'intérêt par analyse d'image
Il existe actuellement deux grandes approches partant de l'analyse d'images des gels 2-DE permettant en premier lieuer la protéomique quantitative. L'une de ces méthodes utilise une comparaison statistique entre plusieurs gels et l'autre utilise un procédé chimique de dérivation des protéines par des sondes fluorescentes donnant la possibilité l'analyse combinée de plusieurs échantillons sur un gel unique. Pour ce faire on utilise dans les deux cas des logiciels d'imagerie. En effet, il est impossible d'appréhender individuellement le nombre énorme de spots (quelquefois jusqu'à 2000) résolues sur un gel 2D (Fig. 5). Ces multiples spots correspondent aux isoformes des protéines scindées en deux dimensions. La position sur le gel des spots polypeptidiques est reproductible dans les dispositifs de séparation en gradient d'Immobilines. Un changement de position est donc un indicateur d'une modification post-traductionelle affectant sa charge et/ou sa taille.
L'analyse d'images repose sur la numérisation de l'image du gel 2-DE après coloration. Au cours de cette étape le logiciel découpe l'image en pixels (contraction de picture element) pour la transmission et le stockage des données. Chaque pixel de l'image est enregistrée à une position en x et en y associée à une valeur de densité optique (DO) proportionnelle à l'intensité du signal enregistré par la caméra ou le scanner. Pour que la DO soit un bon paramètre de mesure et un reflet de l'expression de la protéine, la coloration appliquée doit présenter une gamme dynamique importante et si envisageable linéaire. Dans un gel, en termes de DO le rapport d'intensité entre le plus petit spot détectable et le spot le plus gros est de l'ordre de 104 tandis que la dynamique d'expression des protéines dans la cellule est comprise entre 105 et 106. On constate par conséquent un déficit important de la gamme analytique qui doit être pris en compte au cours de l'analyse.
La coloration des protéines en gel 2-DE reposent essentiellement sur l'emploi de colorants organiques tel que le bleu de Coomassie, de métaux tels que le nitrate d'argent, ou encore par sur des sondes fluorescentes. La gamme de détection fluctue d'un facteur d'environ 10000 entre les méthodes utilisant le bleu de Coomassie (détection de spots contenant une quantité de protéine de l'ordre du µg) et celles utilisant le nitrate d'argent qui permettent d'atteindre 0, 1 ng. Les colorants fluorescents sont moins sensibles que le nitrate d'argent, cependant ont une plus grande reproductibilité et gamme dynamique.
Les logiciels d'analyses d'images actuels incorporent des éléments de visualisation 3D des spots du gel, permettant des changements d'angles en x, y, et z qui sont extrêmement utiles pour séparer des spots proches. La quantification est ainsi perfectionnée. L'utilisation de tels outils permet d'élargir énormément le goulot d'étranglement lié à l'analyse des gels. Cependant, une analyse différentielle fiable nécessite d'établir une comparaison entre des séries d'au moins trois à quatre gels. Des tests statistiques comme l'analyse heuristique ou l'analyse par correspondance permettent objectivement de déterminer la dispersion entre les gels de différentes séries expérimentales.
La multiplication des gels 2D indispensable à l'obtention d'une quantification différentielle statistiquement fiable est cependant un handicap aux analyses à haut débit pour lesquelles la technique du gel unique est intéressante. L'analyse différentielle sur un gel unique : (technologie DIGE pour differential in gel-electrophoresis) a été introduite sur le marché par GE (anciennement) Amersham Biosciences. Le principe repose sur le marquage covalent avec cyanines fluorescentes (p. ex. Cy2, Cy3 et Cy5) des protéines contenues dans deux extraits à analyser. Trois structures sont disponibles avec des spectres de fluorescences différents. Elles possèdent en outre un groupement N-hydroxy-succinimidyl ester qui permet par une réaction de substitution nucléophile avec le groupe amine en epsilon des lysines des protéines de former une amide. L'analyse d'images d'un gel DIGE est plus aisée puisque les deux échantillons ont migré sur le même gel. Les images acquises aux deux longueurs d'onde sont juxtaposées et comparées quantitativement avec logiciels adaptés avec l'ajout d'une référence interne. L'étude comparative en deux couleurs aboutit à la mise en évidence des protéines qui différent ou qui sont semblables dans les deux échantillons. La possibilité offerte d'avoir un étalon interne augmente la fiabilité des mesures quantitatives. Quelle que soit la méthode d'analyse utilisée, les spots d'intérêt une fois détectés sont excisés du gel afin d'être identifiés par des méthodes spectrométriques (spectrométrie de masse en mode MALDI-TOF ou en mode tandem MS/MS). En plus de l'analyse différentielle, l'analyse d'images sert à construire et d'annoter des cartes de référence servant de base à des banques de données consultables sur le WEB.
Identification des protéines
Les différents aspects de l'identification des protéines permettent d'identifier, de caractériser et de quantifier les protéines.
Pour cela :
La masse réelle des protéines peut être mesurée par spectrométrie de masse avec une précision allant de 0, 1 dalton à 10 daltons.
La digestion d'une protéine par une enzyme telle que la trypsine produit des fragments de taille spécifique. La masse des fragments est ensuite mesurée par spectrométrie de masse (technique du fingerprinting).
Par la technique de spectrométrie de masse en tandem et le séquençage d'Edman, il est envisageable de séquencer les peptides.
Identification par spectrométrie de masse (SM)
L'identification par SM repose sur une mesure précise de la masse de peptides ionisés. D'une façon particulièrement générale les protéines sont digérés par une endopeptidase (le plus fréquemment la trypsine) et , ensuite analysées par SM.
Une des approches utilisée est l'établissement de cartes peptidiques massiques («peptide mass fingerprinting»). La masse des peptides obtenus après digestion protéasique est comparée aux cartes de masses théoriques des protéines répertoriées dans les banques de données. Différents algorithmes ont été développés afin d'effectuer cette recherche. Les logiciels d'analyse de données de SM vont rechercher une série de protéines (selon des critères définis par l'expérimentateur) dans une base de données de séquences et générer pour chacune un spectre théorique pour voir lequel se rapproche le plus du spectre expérimental. Selon des logiques différentes pour chaque algorithme, ils établiront un score pour chaque séquence analysée «in silico» qui conduira à un classement des protéines candidates. Cette approche souffre néenmoins de limites objectives et dans le cas d'identifications complexes différents logiciels apporteront des listes de protéines candidates différentes. A titre d'exemple, un logiciel prenant en compte le nombre de masses communes entre le spectre théorique de la protéine candidate et le spectre expérimental facilitera les protéines de haute masse moléculaire pour lesquelles un plus grand nombre de peptides virtuels peuvent être déduits de la séquence.
Une façon de contourner les problèmes rencontrés lors de l'analyse des cartes peptidiques massiques est de séquencer partiellement les protéines par spectrométrie de masse tandem (MS/MS). Dans ce cas, certains fragments peptidiques analysés lors d'une première SM sont choisis et fragmentés. Les pics de masse obtenus forment une représentation de la séquence protéique, dans laquelle deux pics adjacents changent par la masse d'un acide aminé perdu lors de la fragmentation du peptide analysé. La courte séquence protéique obtenue est parfois utilisée pour faire une recherche de ressemblance dans les banques de données. Dans le cas où ces séquences seraient communes à un groupe de protéines, le point isoélectrique et la masse apparente déterminés lors de la séparation par électrophorèse peuvent permettre de trancher. D'une façon générale, le recoupement de plusieurs informations (courtes séquences de quelques acides aminés, localisation d'une fenêtre comprenant le spot en électrophorèse 2D, espèce animale et type cellulaire dont provient l'échantillon) augmente la sécurité pour l'identification d'un polypeptide.
Plutôt que de simplement déterminer une séquence peptidique à partir du spectre de masse d'un peptide et de l'utiliser pour miner une base de données de protéines ou d'ADN, le spectre MS/MS peut être comparé à une série de spectres MS/MS virtuels dérivés des séquences protéiques des bases de données. Partant du même principe, on peut tout d'abord séparer les peptides issus de la digestion trypsique par une nanométhode de chromatographie liquide (nanoLC) et réaliser une MS/MS sur ces différents peptides. La multiplication des informations sur différents segments de la protéine permettra alors non seulement de conforter l'identification, mais également d'obtenir des informations structurales surtout sur les modifications post-traductionnelles, telles que l'addition de groupements phosphates (phosphorylation) ou de chaînes d'oligosaccharides (glycosylation). D'un point de vue fonctionnel, ces informations sont principales. Les phosphorylations sont à la base de la conduction de signaux dans la cellule, de l'extérieur de celle-ci par ses récepteurs membranaires jusqu'au noyau où sont centralisées les informations régulant la vie cellulaire. Quant aux chaînes oligosaccharidiques, elles jouent un rôle essentiel dans la modulation des propriétés chimiques de certaines protéines (glycoprotéines) et gouvernent quelquefois leur activité biologique. Pour identifier ces groupements, la SM utilisera les fragments résultant de leur séparation de la chaîne peptidique ou de leur propre fragmentation.
Interrogation des bases de données
La dernière étape est l'interrogation des bases de données. Les différentes informations récoltées sur les protéines (masse apparente, masse réelle, point isoélectrique, taille des fragments après digestion enzymatique, séquence partielles) sont comparées aux bases de données génomiques ou protéomiques en ligne. Les logiciels fournissent alors une liste de protéines et leurs probabilités associées.
Cette obligation de passer par les bases de données explique l'importance de la bio-informatique pour le protéomicien.
Corganisme dont le génome n'a pas encore été séquencé, Seuls certains organismes, dénommé organisme modèle, ont leurs génomes totalement séquencés et disponibles en ligne. Dans le cas opposé, les organismes sont étudiés par homologie avec les organismes connus.
Quantification de l'expression des protéines
Elle sert à quantifier les variations de leurs taux d'expression selon le temps, de leurs environnements, de leurs états de développement, de leurs états physiologiques et pathologiques, de l'espèce d'origine.
Les techniques les plus fréquemment utilisées sont :
Les interactions protéiques
Elle étudie aussi les interactions que les protéines ont avec d'autres protéines, avec l'ADN ou l'ARN, avec des substances.
Une autre manière de considérer les interactions protéiques est de faire du double hybride. En criblant une banque d'ADNc avec sa protéine comme appât on peut déceler l'ensemble des interractants dans un organisme ou un tissu spécifique (animal ou végétal). Correctement utilisée, cette technique est particulièrement efficace. La qualité des résultats dépend fréquemment de la qualité de la banque et de l'efficacité de la transformation de la levure ou de la bactérie.
Protéomique fonctionnelle
La protéomique fonctionnelle étudie les fonctions de chaque protéine.
Protéomique structurale
La protéomique étudie enfin les structures primaires, secondaires et tertiaires des protéines.
Les enjeux de la protéomique
La recherche de biomarqueurs
De nouveaux outils thérapeutiques
Notes et références
- ↑ P. James, «Protein identification in the post-genome era : the rapid rise of proteomics. », dans Quarterly reviews of biophysics, vol. 30, no 4, 1997, p. 279–331
- ↑ http ://www. nobel-prize. org/FR
Voir aussi
Bibliographie
- Greg Gibson, Spencer V. Muse, Précis de génomique, Editions De Bœck Université, 2004, ISBN 2804143341.
- Reiner Westermeier, Tom Naven, Protéomics in practice, Wiley-VCH, 2002, ISBN 3527303545.
- Thierry Rabilloud, Proteome reserch : Two-dimensionnal gel electrophoresis an identification methods, Springer Verlag Berlin Heidelberg, 2000, ISBN 3540657924
- Marc R. Wilkins, Keith L. Williams, Ron D. Appel, Denis F. Hochstrasser, Proteome research : New frontiers in functional genomics, Springer Verlag Berlin Heidelberg, 1997, ISBN 3540627537.
Liens externes
- Société Française d'électrophorèse et d'analyse protéomique Congrès, bourses, electrophorum
- Société de BioChromatographie et Nanoséparations Réunions scientifiques, informations. S'intéresse surtout à la séparation des peptides et protéines par chromatographie ou par micro- et nano-méthodes
- Site du master protéomique de Lille Actualité scientifique, dates de congrès, présentation du master.
- Hybrigenics : Site d'une société faisant du double hybride Société faisant du double hybride et servant à trouver des interractions protéiques pour une protéine appat donnée.
- The state of the art in the analysis of two-dimensional gel electrophoresis images
Recherche sur Amazone (livres) : |
Voir la liste des contributeurs.
La version présentée ici à été extraite depuis cette source le 05/11/2009.
Ce texte est disponible sous les termes de la licence de documentation libre GNU (GFDL).
La liste des définitions proposées en tête de page est une sélection parmi les résultats obtenus à l'aide de la commande "define:" de Google.
Cette page fait partie du projet Wikibis.
 Accueil
Accueil Recherche
Recherche Début page
Début page Contact
Contact Imprimer
Imprimer Accessibilité
Accessibilité